Content from Introduction and setup
Last updated on 2023-03-14 | Edit this page
Overview
Questions
Objectives
- Install required packages.
- Download the data.
Download data
The data we will use in this lesson is obtained from the Gene Expression Omnibus, accession number GSE96870.
R
dir.create("data", showWarnings = FALSE)
download.file(
url = "https://github.com/Bioconductor/bioconductor-teaching/blob/master/data/GSE96870/GSE96870_counts_cerebellum.csv?raw=true",
destfile = "data/GSE96870_counts_cerebellum.csv"
)
download.file(
url = "https://github.com/Bioconductor/bioconductor-teaching/blob/master/data/GSE96870/GSE96870_coldata_cerebellum.csv?raw=true",
destfile = "data/GSE96870_coldata_cerebellum.csv"
)
download.file(
url = "https://github.com/Bioconductor/bioconductor-teaching/blob/master/data/GSE96870/GSE96870_coldata_all.csv?raw=true",
destfile = "data/GSE96870_coldata_all.csv"
)
download.file(
url = "https://github.com/Bioconductor/bioconductor-teaching/blob/master/data/GSE96870/GSE96870_rowranges.tsv?raw=true",
destfile = "data/GSE96870_rowranges.tsv"
)
Content from Introduction to RNA-seq
Last updated on 2023-03-14 | Edit this page
Overview
Questions
- What is RNA-seq?
Objectives
- Explain what RNA-seq is and what the generated data looks like.
- Provide an overview of common quality control steps for the raw data.
- Explain how gene expression levels can be estimated from raw data.
Content from Experimental design
Last updated on 2023-03-14 | Edit this page
Overview
Questions
- How do we design experiments optimally?
- How do we interpret a given design?
Objectives
- Explain the formula notation and design matrices.
- Explore different designs and learn how to interpret coefficients.
R
suppressPackageStartupMessages({
library(SummarizedExperiment)
library(ExploreModelMatrix)
library(dplyr)
})
R
meta <- read.csv("data/GSE96870_coldata_all.csv", row.names = 1)
meta
OUTPUT
title geo_accession organism age sex
GSM2545336 CNS_RNA-seq_10C GSM2545336 Mus musculus 8 weeks Female
GSM2545337 CNS_RNA-seq_11C GSM2545337 Mus musculus 8 weeks Female
GSM2545338 CNS_RNA-seq_12C GSM2545338 Mus musculus 8 weeks Female
GSM2545339 CNS_RNA-seq_13C GSM2545339 Mus musculus 8 weeks Female
GSM2545340 CNS_RNA-seq_14C GSM2545340 Mus musculus 8 weeks Male
GSM2545341 CNS_RNA-seq_17C GSM2545341 Mus musculus 8 weeks Male
GSM2545342 CNS_RNA-seq_1C GSM2545342 Mus musculus 8 weeks Female
GSM2545343 CNS_RNA-seq_20C GSM2545343 Mus musculus 8 weeks Male
GSM2545344 CNS_RNA-seq_21C GSM2545344 Mus musculus 8 weeks Female
GSM2545345 CNS_RNA-seq_22C GSM2545345 Mus musculus 8 weeks Male
GSM2545346 CNS_RNA-seq_25C GSM2545346 Mus musculus 8 weeks Male
GSM2545347 CNS_RNA-seq_26C GSM2545347 Mus musculus 8 weeks Male
GSM2545348 CNS_RNA-seq_27C GSM2545348 Mus musculus 8 weeks Female
GSM2545349 CNS_RNA-seq_28C GSM2545349 Mus musculus 8 weeks Male
GSM2545350 CNS_RNA-seq_29C GSM2545350 Mus musculus 8 weeks Male
GSM2545351 CNS_RNA-seq_2C GSM2545351 Mus musculus 8 weeks Female
GSM2545352 CNS_RNA-seq_30C GSM2545352 Mus musculus 8 weeks Female
GSM2545353 CNS_RNA-seq_3C GSM2545353 Mus musculus 8 weeks Female
GSM2545354 CNS_RNA-seq_4C GSM2545354 Mus musculus 8 weeks Male
GSM2545355 CNS_RNA-seq_571 GSM2545355 Mus musculus 8 weeks Male
GSM2545356 CNS_RNA-seq_574 GSM2545356 Mus musculus 8 weeks Male
GSM2545357 CNS_RNA-seq_575 GSM2545357 Mus musculus 8 weeks Male
GSM2545358 CNS_RNA-seq_583 GSM2545358 Mus musculus 8 weeks Female
GSM2545359 CNS_RNA-seq_585 GSM2545359 Mus musculus 8 weeks Female
GSM2545360 CNS_RNA-seq_589 GSM2545360 Mus musculus 8 weeks Male
GSM2545361 CNS_RNA-seq_590 GSM2545361 Mus musculus 8 weeks Male
GSM2545362 CNS_RNA-seq_5C GSM2545362 Mus musculus 8 weeks Female
GSM2545363 CNS_RNA-seq_6C GSM2545363 Mus musculus 8 weeks Male
GSM2545364 CNS_RNA-seq_709 GSM2545364 Mus musculus 8 weeks Female
GSM2545365 CNS_RNA-seq_710 GSM2545365 Mus musculus 8 weeks Female
GSM2545366 CNS_RNA-seq_711 GSM2545366 Mus musculus 8 weeks Female
GSM2545367 CNS_RNA-seq_713 GSM2545367 Mus musculus 8 weeks Male
GSM2545368 CNS_RNA-seq_728 GSM2545368 Mus musculus 8 weeks Male
GSM2545369 CNS_RNA-seq_729 GSM2545369 Mus musculus 8 weeks Male
GSM2545370 CNS_RNA-seq_730 GSM2545370 Mus musculus 8 weeks Female
GSM2545371 CNS_RNA-seq_731 GSM2545371 Mus musculus 8 weeks Female
GSM2545372 CNS_RNA-seq_733 GSM2545372 Mus musculus 8 weeks Male
GSM2545373 CNS_RNA-seq_735 GSM2545373 Mus musculus 8 weeks Male
GSM2545374 CNS_RNA-seq_736 GSM2545374 Mus musculus 8 weeks Female
GSM2545375 CNS_RNA-seq_738 GSM2545375 Mus musculus 8 weeks Female
GSM2545376 CNS_RNA-seq_740 GSM2545376 Mus musculus 8 weeks Female
GSM2545377 CNS_RNA-seq_741 GSM2545377 Mus musculus 8 weeks Female
GSM2545378 CNS_RNA-seq_742 GSM2545378 Mus musculus 8 weeks Male
GSM2545379 CNS_RNA-seq_743 GSM2545379 Mus musculus 8 weeks Male
GSM2545380 CNS_RNA-seq_9C GSM2545380 Mus musculus 8 weeks Female
infection strain time tissue mouse
GSM2545336 InfluenzaA C57BL/6 Day8 Cerebellum 14
GSM2545337 NonInfected C57BL/6 Day0 Cerebellum 9
GSM2545338 NonInfected C57BL/6 Day0 Cerebellum 10
GSM2545339 InfluenzaA C57BL/6 Day4 Cerebellum 15
GSM2545340 InfluenzaA C57BL/6 Day4 Cerebellum 18
GSM2545341 InfluenzaA C57BL/6 Day8 Cerebellum 6
GSM2545342 InfluenzaA C57BL/6 Day8 Cerebellum 5
GSM2545343 NonInfected C57BL/6 Day0 Cerebellum 11
GSM2545344 InfluenzaA C57BL/6 Day4 Cerebellum 22
GSM2545345 InfluenzaA C57BL/6 Day4 Cerebellum 13
GSM2545346 InfluenzaA C57BL/6 Day8 Cerebellum 23
GSM2545347 InfluenzaA C57BL/6 Day8 Cerebellum 24
GSM2545348 NonInfected C57BL/6 Day0 Cerebellum 8
GSM2545349 NonInfected C57BL/6 Day0 Cerebellum 7
GSM2545350 InfluenzaA C57BL/6 Day4 Cerebellum 1
GSM2545351 InfluenzaA C57BL/6 Day8 Cerebellum 16
GSM2545352 InfluenzaA C57BL/6 Day4 Cerebellum 21
GSM2545353 NonInfected C57BL/6 Day0 Cerebellum 4
GSM2545354 NonInfected C57BL/6 Day0 Cerebellum 2
GSM2545355 InfluenzaA C57BL/6 Day4 Spinalcord 1
GSM2545356 NonInfected C57BL/6 Day0 Spinalcord 2
GSM2545357 NonInfected C57BL/6 Day0 Spinalcord 3
GSM2545358 NonInfected C57BL/6 Day0 Spinalcord 4
GSM2545359 InfluenzaA C57BL/6 Day8 Spinalcord 5
GSM2545360 InfluenzaA C57BL/6 Day8 Spinalcord 6
GSM2545361 NonInfected C57BL/6 Day0 Spinalcord 7
GSM2545362 InfluenzaA C57BL/6 Day4 Cerebellum 20
GSM2545363 InfluenzaA C57BL/6 Day4 Cerebellum 12
GSM2545364 NonInfected C57BL/6 Day0 Spinalcord 8
GSM2545365 NonInfected C57BL/6 Day0 Spinalcord 9
GSM2545366 NonInfected C57BL/6 Day0 Spinalcord 10
GSM2545367 NonInfected C57BL/6 Day0 Spinalcord 11
GSM2545368 InfluenzaA C57BL/6 Day4 Spinalcord 12
GSM2545369 InfluenzaA C57BL/6 Day4 Spinalcord 13
GSM2545370 InfluenzaA C57BL/6 Day8 Spinalcord 14
GSM2545371 InfluenzaA C57BL/6 Day4 Spinalcord 15
GSM2545372 InfluenzaA C57BL/6 Day8 Spinalcord 17
GSM2545373 InfluenzaA C57BL/6 Day4 Spinalcord 18
GSM2545374 InfluenzaA C57BL/6 Day8 Spinalcord 19
GSM2545375 InfluenzaA C57BL/6 Day4 Spinalcord 20
GSM2545376 InfluenzaA C57BL/6 Day4 Spinalcord 21
GSM2545377 InfluenzaA C57BL/6 Day4 Spinalcord 22
GSM2545378 InfluenzaA C57BL/6 Day8 Spinalcord 23
GSM2545379 InfluenzaA C57BL/6 Day8 Spinalcord 24
GSM2545380 InfluenzaA C57BL/6 Day8 Cerebellum 19R
vd <- VisualizeDesign(sampleData = meta,
designFormula = ~ tissue + time + sex)
vd$cooccurrenceplots
OUTPUT
$`tissue = Cerebellum`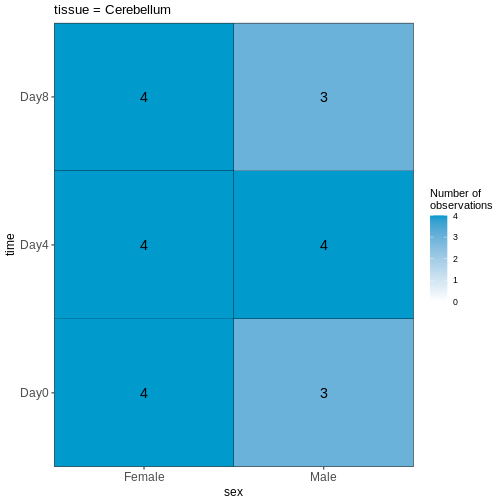
OUTPUT
$`tissue = Spinalcord`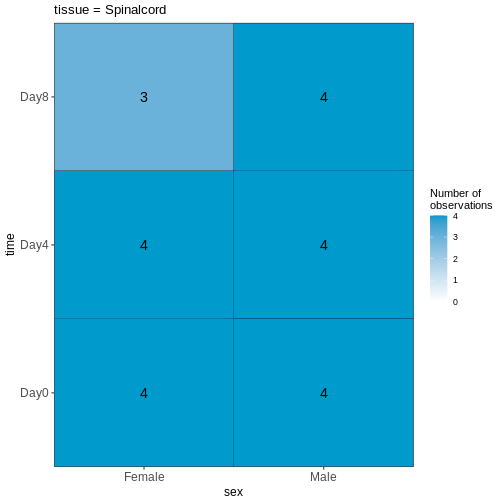
Compare males and females, non-infected spinal cord
R
meta_noninf_spc <- meta %>% filter(time == "Day0" &
tissue == "Spinalcord")
meta_noninf_spc
OUTPUT
title geo_accession organism age sex
GSM2545356 CNS_RNA-seq_574 GSM2545356 Mus musculus 8 weeks Male
GSM2545357 CNS_RNA-seq_575 GSM2545357 Mus musculus 8 weeks Male
GSM2545358 CNS_RNA-seq_583 GSM2545358 Mus musculus 8 weeks Female
GSM2545361 CNS_RNA-seq_590 GSM2545361 Mus musculus 8 weeks Male
GSM2545364 CNS_RNA-seq_709 GSM2545364 Mus musculus 8 weeks Female
GSM2545365 CNS_RNA-seq_710 GSM2545365 Mus musculus 8 weeks Female
GSM2545366 CNS_RNA-seq_711 GSM2545366 Mus musculus 8 weeks Female
GSM2545367 CNS_RNA-seq_713 GSM2545367 Mus musculus 8 weeks Male
infection strain time tissue mouse
GSM2545356 NonInfected C57BL/6 Day0 Spinalcord 2
GSM2545357 NonInfected C57BL/6 Day0 Spinalcord 3
GSM2545358 NonInfected C57BL/6 Day0 Spinalcord 4
GSM2545361 NonInfected C57BL/6 Day0 Spinalcord 7
GSM2545364 NonInfected C57BL/6 Day0 Spinalcord 8
GSM2545365 NonInfected C57BL/6 Day0 Spinalcord 9
GSM2545366 NonInfected C57BL/6 Day0 Spinalcord 10
GSM2545367 NonInfected C57BL/6 Day0 Spinalcord 11R
vd <- VisualizeDesign(sampleData = meta_noninf_spc,
designFormula = ~ sex)
vd$designmatrix
OUTPUT
(Intercept) sexMale
GSM2545356 1 1
GSM2545357 1 1
GSM2545358 1 0
GSM2545361 1 1
GSM2545364 1 0
GSM2545365 1 0
GSM2545366 1 0
GSM2545367 1 1R
vd$plotlist
OUTPUT
[[1]]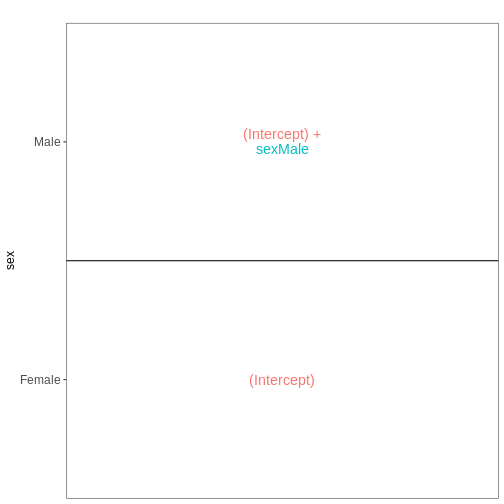
R
meta_male_spc <- meta %>% filter(sex == "Male" & tissue == "Spinalcord")
meta_male_spc
OUTPUT
title geo_accession organism age sex infection
GSM2545355 CNS_RNA-seq_571 GSM2545355 Mus musculus 8 weeks Male InfluenzaA
GSM2545356 CNS_RNA-seq_574 GSM2545356 Mus musculus 8 weeks Male NonInfected
GSM2545357 CNS_RNA-seq_575 GSM2545357 Mus musculus 8 weeks Male NonInfected
GSM2545360 CNS_RNA-seq_589 GSM2545360 Mus musculus 8 weeks Male InfluenzaA
GSM2545361 CNS_RNA-seq_590 GSM2545361 Mus musculus 8 weeks Male NonInfected
GSM2545367 CNS_RNA-seq_713 GSM2545367 Mus musculus 8 weeks Male NonInfected
GSM2545368 CNS_RNA-seq_728 GSM2545368 Mus musculus 8 weeks Male InfluenzaA
GSM2545369 CNS_RNA-seq_729 GSM2545369 Mus musculus 8 weeks Male InfluenzaA
GSM2545372 CNS_RNA-seq_733 GSM2545372 Mus musculus 8 weeks Male InfluenzaA
GSM2545373 CNS_RNA-seq_735 GSM2545373 Mus musculus 8 weeks Male InfluenzaA
GSM2545378 CNS_RNA-seq_742 GSM2545378 Mus musculus 8 weeks Male InfluenzaA
GSM2545379 CNS_RNA-seq_743 GSM2545379 Mus musculus 8 weeks Male InfluenzaA
strain time tissue mouse
GSM2545355 C57BL/6 Day4 Spinalcord 1
GSM2545356 C57BL/6 Day0 Spinalcord 2
GSM2545357 C57BL/6 Day0 Spinalcord 3
GSM2545360 C57BL/6 Day8 Spinalcord 6
GSM2545361 C57BL/6 Day0 Spinalcord 7
GSM2545367 C57BL/6 Day0 Spinalcord 11
GSM2545368 C57BL/6 Day4 Spinalcord 12
GSM2545369 C57BL/6 Day4 Spinalcord 13
GSM2545372 C57BL/6 Day8 Spinalcord 17
GSM2545373 C57BL/6 Day4 Spinalcord 18
GSM2545378 C57BL/6 Day8 Spinalcord 23
GSM2545379 C57BL/6 Day8 Spinalcord 24R
vd <- VisualizeDesign(sampleData = meta_male_spc, designFormula = ~ time)
vd$designmatrix
OUTPUT
(Intercept) timeDay4 timeDay8
GSM2545355 1 1 0
GSM2545356 1 0 0
GSM2545357 1 0 0
GSM2545360 1 0 1
GSM2545361 1 0 0
GSM2545367 1 0 0
GSM2545368 1 1 0
GSM2545369 1 1 0
GSM2545372 1 0 1
GSM2545373 1 1 0
GSM2545378 1 0 1
GSM2545379 1 0 1R
vd$plotlist
OUTPUT
[[1]]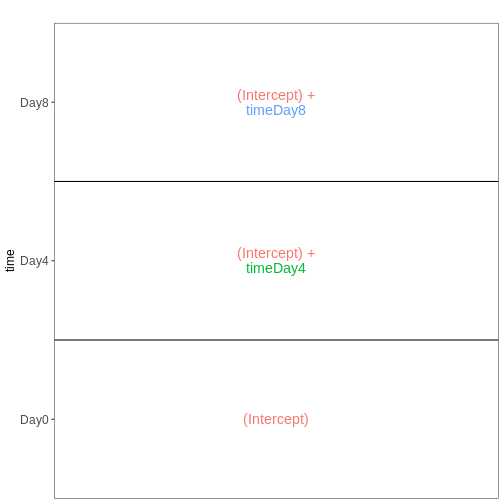
Factorial design without interactions
R
meta_noninf <- meta %>% filter(time == "Day0")
meta_noninf
OUTPUT
title geo_accession organism age sex
GSM2545337 CNS_RNA-seq_11C GSM2545337 Mus musculus 8 weeks Female
GSM2545338 CNS_RNA-seq_12C GSM2545338 Mus musculus 8 weeks Female
GSM2545343 CNS_RNA-seq_20C GSM2545343 Mus musculus 8 weeks Male
GSM2545348 CNS_RNA-seq_27C GSM2545348 Mus musculus 8 weeks Female
GSM2545349 CNS_RNA-seq_28C GSM2545349 Mus musculus 8 weeks Male
GSM2545353 CNS_RNA-seq_3C GSM2545353 Mus musculus 8 weeks Female
GSM2545354 CNS_RNA-seq_4C GSM2545354 Mus musculus 8 weeks Male
GSM2545356 CNS_RNA-seq_574 GSM2545356 Mus musculus 8 weeks Male
GSM2545357 CNS_RNA-seq_575 GSM2545357 Mus musculus 8 weeks Male
GSM2545358 CNS_RNA-seq_583 GSM2545358 Mus musculus 8 weeks Female
GSM2545361 CNS_RNA-seq_590 GSM2545361 Mus musculus 8 weeks Male
GSM2545364 CNS_RNA-seq_709 GSM2545364 Mus musculus 8 weeks Female
GSM2545365 CNS_RNA-seq_710 GSM2545365 Mus musculus 8 weeks Female
GSM2545366 CNS_RNA-seq_711 GSM2545366 Mus musculus 8 weeks Female
GSM2545367 CNS_RNA-seq_713 GSM2545367 Mus musculus 8 weeks Male
infection strain time tissue mouse
GSM2545337 NonInfected C57BL/6 Day0 Cerebellum 9
GSM2545338 NonInfected C57BL/6 Day0 Cerebellum 10
GSM2545343 NonInfected C57BL/6 Day0 Cerebellum 11
GSM2545348 NonInfected C57BL/6 Day0 Cerebellum 8
GSM2545349 NonInfected C57BL/6 Day0 Cerebellum 7
GSM2545353 NonInfected C57BL/6 Day0 Cerebellum 4
GSM2545354 NonInfected C57BL/6 Day0 Cerebellum 2
GSM2545356 NonInfected C57BL/6 Day0 Spinalcord 2
GSM2545357 NonInfected C57BL/6 Day0 Spinalcord 3
GSM2545358 NonInfected C57BL/6 Day0 Spinalcord 4
GSM2545361 NonInfected C57BL/6 Day0 Spinalcord 7
GSM2545364 NonInfected C57BL/6 Day0 Spinalcord 8
GSM2545365 NonInfected C57BL/6 Day0 Spinalcord 9
GSM2545366 NonInfected C57BL/6 Day0 Spinalcord 10
GSM2545367 NonInfected C57BL/6 Day0 Spinalcord 11R
vd <- VisualizeDesign(sampleData = meta_noninf,
designFormula = ~ sex + tissue)
vd$designmatrix
OUTPUT
(Intercept) sexMale tissueSpinalcord
GSM2545337 1 0 0
GSM2545338 1 0 0
GSM2545343 1 1 0
GSM2545348 1 0 0
GSM2545349 1 1 0
GSM2545353 1 0 0
GSM2545354 1 1 0
GSM2545356 1 1 1
GSM2545357 1 1 1
GSM2545358 1 0 1
GSM2545361 1 1 1
GSM2545364 1 0 1
GSM2545365 1 0 1
GSM2545366 1 0 1
GSM2545367 1 1 1R
vd$plotlist
OUTPUT
[[1]]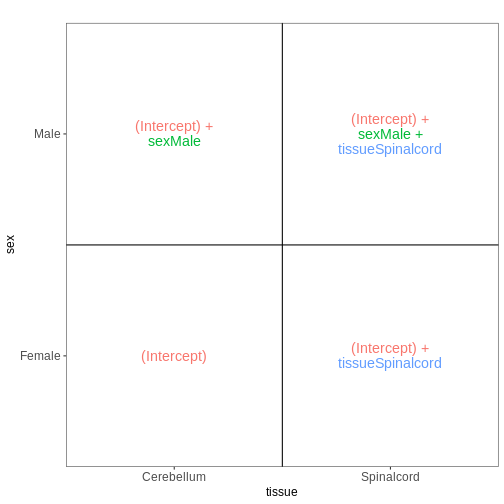
Factorial design with interactions
R
meta_noninf <- meta %>% filter(time == "Day0")
meta_noninf
OUTPUT
title geo_accession organism age sex
GSM2545337 CNS_RNA-seq_11C GSM2545337 Mus musculus 8 weeks Female
GSM2545338 CNS_RNA-seq_12C GSM2545338 Mus musculus 8 weeks Female
GSM2545343 CNS_RNA-seq_20C GSM2545343 Mus musculus 8 weeks Male
GSM2545348 CNS_RNA-seq_27C GSM2545348 Mus musculus 8 weeks Female
GSM2545349 CNS_RNA-seq_28C GSM2545349 Mus musculus 8 weeks Male
GSM2545353 CNS_RNA-seq_3C GSM2545353 Mus musculus 8 weeks Female
GSM2545354 CNS_RNA-seq_4C GSM2545354 Mus musculus 8 weeks Male
GSM2545356 CNS_RNA-seq_574 GSM2545356 Mus musculus 8 weeks Male
GSM2545357 CNS_RNA-seq_575 GSM2545357 Mus musculus 8 weeks Male
GSM2545358 CNS_RNA-seq_583 GSM2545358 Mus musculus 8 weeks Female
GSM2545361 CNS_RNA-seq_590 GSM2545361 Mus musculus 8 weeks Male
GSM2545364 CNS_RNA-seq_709 GSM2545364 Mus musculus 8 weeks Female
GSM2545365 CNS_RNA-seq_710 GSM2545365 Mus musculus 8 weeks Female
GSM2545366 CNS_RNA-seq_711 GSM2545366 Mus musculus 8 weeks Female
GSM2545367 CNS_RNA-seq_713 GSM2545367 Mus musculus 8 weeks Male
infection strain time tissue mouse
GSM2545337 NonInfected C57BL/6 Day0 Cerebellum 9
GSM2545338 NonInfected C57BL/6 Day0 Cerebellum 10
GSM2545343 NonInfected C57BL/6 Day0 Cerebellum 11
GSM2545348 NonInfected C57BL/6 Day0 Cerebellum 8
GSM2545349 NonInfected C57BL/6 Day0 Cerebellum 7
GSM2545353 NonInfected C57BL/6 Day0 Cerebellum 4
GSM2545354 NonInfected C57BL/6 Day0 Cerebellum 2
GSM2545356 NonInfected C57BL/6 Day0 Spinalcord 2
GSM2545357 NonInfected C57BL/6 Day0 Spinalcord 3
GSM2545358 NonInfected C57BL/6 Day0 Spinalcord 4
GSM2545361 NonInfected C57BL/6 Day0 Spinalcord 7
GSM2545364 NonInfected C57BL/6 Day0 Spinalcord 8
GSM2545365 NonInfected C57BL/6 Day0 Spinalcord 9
GSM2545366 NonInfected C57BL/6 Day0 Spinalcord 10
GSM2545367 NonInfected C57BL/6 Day0 Spinalcord 11R
vd <- VisualizeDesign(sampleData = meta_noninf,
designFormula = ~ sex * tissue)
vd$designmatrix
OUTPUT
(Intercept) sexMale tissueSpinalcord sexMale:tissueSpinalcord
GSM2545337 1 0 0 0
GSM2545338 1 0 0 0
GSM2545343 1 1 0 0
GSM2545348 1 0 0 0
GSM2545349 1 1 0 0
GSM2545353 1 0 0 0
GSM2545354 1 1 0 0
GSM2545356 1 1 1 1
GSM2545357 1 1 1 1
GSM2545358 1 0 1 0
GSM2545361 1 1 1 1
GSM2545364 1 0 1 0
GSM2545365 1 0 1 0
GSM2545366 1 0 1 0
GSM2545367 1 1 1 1R
vd$plotlist
OUTPUT
[[1]]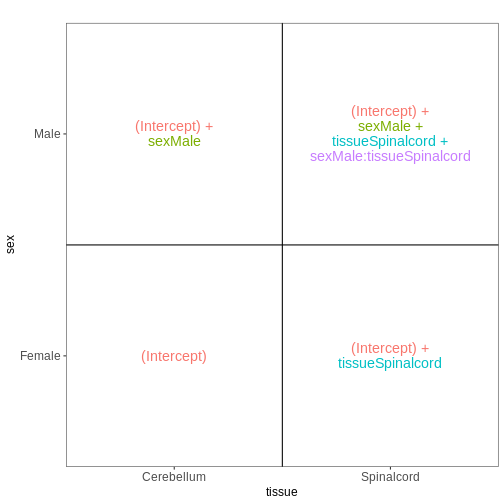
Paired design
R
meta_fem_day0 <- meta %>% filter(sex == "Female" &
time == "Day0")
meta_fem_day0
OUTPUT
title geo_accession organism age sex
GSM2545337 CNS_RNA-seq_11C GSM2545337 Mus musculus 8 weeks Female
GSM2545338 CNS_RNA-seq_12C GSM2545338 Mus musculus 8 weeks Female
GSM2545348 CNS_RNA-seq_27C GSM2545348 Mus musculus 8 weeks Female
GSM2545353 CNS_RNA-seq_3C GSM2545353 Mus musculus 8 weeks Female
GSM2545358 CNS_RNA-seq_583 GSM2545358 Mus musculus 8 weeks Female
GSM2545364 CNS_RNA-seq_709 GSM2545364 Mus musculus 8 weeks Female
GSM2545365 CNS_RNA-seq_710 GSM2545365 Mus musculus 8 weeks Female
GSM2545366 CNS_RNA-seq_711 GSM2545366 Mus musculus 8 weeks Female
infection strain time tissue mouse
GSM2545337 NonInfected C57BL/6 Day0 Cerebellum 9
GSM2545338 NonInfected C57BL/6 Day0 Cerebellum 10
GSM2545348 NonInfected C57BL/6 Day0 Cerebellum 8
GSM2545353 NonInfected C57BL/6 Day0 Cerebellum 4
GSM2545358 NonInfected C57BL/6 Day0 Spinalcord 4
GSM2545364 NonInfected C57BL/6 Day0 Spinalcord 8
GSM2545365 NonInfected C57BL/6 Day0 Spinalcord 9
GSM2545366 NonInfected C57BL/6 Day0 Spinalcord 10R
vd <- VisualizeDesign(sampleData = meta_fem_day0,
designFormula = ~ mouse + tissue)
vd$designmatrix
OUTPUT
(Intercept) mouse tissueSpinalcord
GSM2545337 1 9 0
GSM2545338 1 10 0
GSM2545348 1 8 0
GSM2545353 1 4 0
GSM2545358 1 4 1
GSM2545364 1 8 1
GSM2545365 1 9 1
GSM2545366 1 10 1R
vd$plotlist
OUTPUT
[[1]]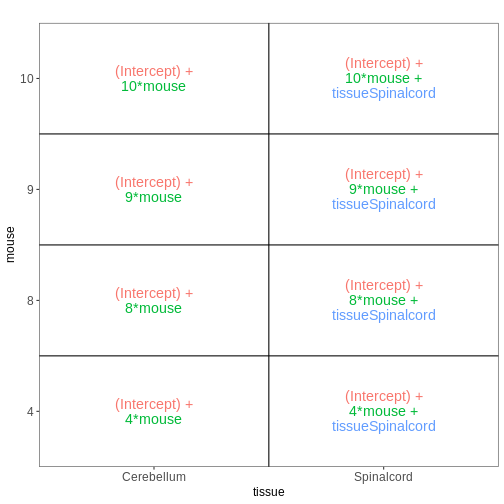
Within- and between-subject comparisons
R
meta_fem_day04 <- meta %>%
filter(sex == "Female" &
time %in% c("Day0", "Day4")) %>%
droplevels()
meta_fem_day04
OUTPUT
title geo_accession organism age sex
GSM2545337 CNS_RNA-seq_11C GSM2545337 Mus musculus 8 weeks Female
GSM2545338 CNS_RNA-seq_12C GSM2545338 Mus musculus 8 weeks Female
GSM2545339 CNS_RNA-seq_13C GSM2545339 Mus musculus 8 weeks Female
GSM2545344 CNS_RNA-seq_21C GSM2545344 Mus musculus 8 weeks Female
GSM2545348 CNS_RNA-seq_27C GSM2545348 Mus musculus 8 weeks Female
GSM2545352 CNS_RNA-seq_30C GSM2545352 Mus musculus 8 weeks Female
GSM2545353 CNS_RNA-seq_3C GSM2545353 Mus musculus 8 weeks Female
GSM2545358 CNS_RNA-seq_583 GSM2545358 Mus musculus 8 weeks Female
GSM2545362 CNS_RNA-seq_5C GSM2545362 Mus musculus 8 weeks Female
GSM2545364 CNS_RNA-seq_709 GSM2545364 Mus musculus 8 weeks Female
GSM2545365 CNS_RNA-seq_710 GSM2545365 Mus musculus 8 weeks Female
GSM2545366 CNS_RNA-seq_711 GSM2545366 Mus musculus 8 weeks Female
GSM2545371 CNS_RNA-seq_731 GSM2545371 Mus musculus 8 weeks Female
GSM2545375 CNS_RNA-seq_738 GSM2545375 Mus musculus 8 weeks Female
GSM2545376 CNS_RNA-seq_740 GSM2545376 Mus musculus 8 weeks Female
GSM2545377 CNS_RNA-seq_741 GSM2545377 Mus musculus 8 weeks Female
infection strain time tissue mouse
GSM2545337 NonInfected C57BL/6 Day0 Cerebellum 9
GSM2545338 NonInfected C57BL/6 Day0 Cerebellum 10
GSM2545339 InfluenzaA C57BL/6 Day4 Cerebellum 15
GSM2545344 InfluenzaA C57BL/6 Day4 Cerebellum 22
GSM2545348 NonInfected C57BL/6 Day0 Cerebellum 8
GSM2545352 InfluenzaA C57BL/6 Day4 Cerebellum 21
GSM2545353 NonInfected C57BL/6 Day0 Cerebellum 4
GSM2545358 NonInfected C57BL/6 Day0 Spinalcord 4
GSM2545362 InfluenzaA C57BL/6 Day4 Cerebellum 20
GSM2545364 NonInfected C57BL/6 Day0 Spinalcord 8
GSM2545365 NonInfected C57BL/6 Day0 Spinalcord 9
GSM2545366 NonInfected C57BL/6 Day0 Spinalcord 10
GSM2545371 InfluenzaA C57BL/6 Day4 Spinalcord 15
GSM2545375 InfluenzaA C57BL/6 Day4 Spinalcord 20
GSM2545376 InfluenzaA C57BL/6 Day4 Spinalcord 21
GSM2545377 InfluenzaA C57BL/6 Day4 Spinalcord 22R
design <- model.matrix(~ mouse, data = meta_fem_day04)
design <- cbind(design,
Spc.Day0 = meta_fem_day04$tissue == "Spinalcord" &
meta_fem_day04$time == "Day0",
Spc.Day4 = meta_fem_day04$tissue == "Spinalcord" &
meta_fem_day04$time == "Day4")
rownames(design) <- rownames(meta_fem_day04)
design
OUTPUT
(Intercept) mouse Spc.Day0 Spc.Day4
GSM2545337 1 9 0 0
GSM2545338 1 10 0 0
GSM2545339 1 15 0 0
GSM2545344 1 22 0 0
GSM2545348 1 8 0 0
GSM2545352 1 21 0 0
GSM2545353 1 4 0 0
GSM2545358 1 4 1 0
GSM2545362 1 20 0 0
GSM2545364 1 8 1 0
GSM2545365 1 9 1 0
GSM2545366 1 10 1 0
GSM2545371 1 15 0 1
GSM2545375 1 20 0 1
GSM2545376 1 21 0 1
GSM2545377 1 22 0 1R
vd <- VisualizeDesign(sampleData = meta_fem_day04 %>%
select(time, tissue, mouse),
designFormula = NULL,
designMatrix = design, flipCoordFitted = FALSE)
vd$designmatrix
OUTPUT
(Intercept) mouse Spc.Day0 Spc.Day4
GSM2545337 1 9 0 0
GSM2545338 1 10 0 0
GSM2545339 1 15 0 0
GSM2545344 1 22 0 0
GSM2545348 1 8 0 0
GSM2545352 1 21 0 0
GSM2545353 1 4 0 0
GSM2545358 1 4 1 0
GSM2545362 1 20 0 0
GSM2545364 1 8 1 0
GSM2545365 1 9 1 0
GSM2545366 1 10 1 0
GSM2545371 1 15 0 1
GSM2545375 1 20 0 1
GSM2545376 1 21 0 1
GSM2545377 1 22 0 1R
vd$plotlist
OUTPUT
$`time = Day0`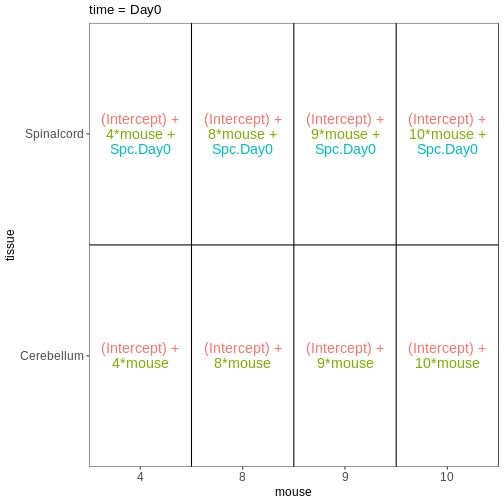
OUTPUT
$`time = Day4`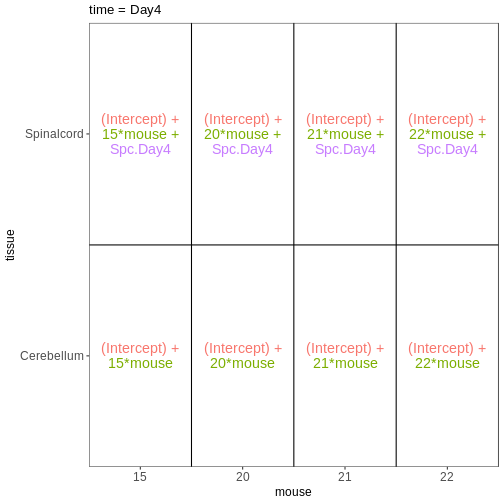
Content from Importing and annotating quantified data into R
Last updated on 2023-03-14 | Edit this page
Overview
Questions
- How do we get our data into R?
Objectives
- Learn how to import the quantifications into a SummarizedExperiment object.
- Learn how to add additional gene annotations to the object.
Contribute!
This episode is intended to show how we can assemble a SummarizedExperiment starting from individual count, rowdata and coldata files. Moreover, we will practice adding annotations for the genes, and discuss related concepts and things to keep in mind (annotation sources, versions, ‘helper’ packages like tximeta).
Read the data
Gene annotations
Need to be careful - the descriptions contain both commas and ’ (e.g., 5’)
R
rowranges <- read.delim("data/GSE96870_rowranges.tsv", sep = "\t",
colClasses = c(ENTREZID = "character"),
header = TRUE, quote = "", row.names = 5)
Mention other ways of getting annotations, and practice querying org package. Important to use the right annotation source/version.
R
suppressPackageStartupMessages({
library(org.Mm.eg.db)
})
mapIds(org.Mm.eg.db, keys = "497097", column = "SYMBOL", keytype = "ENTREZID")
OUTPUT
'select()' returned 1:1 mapping between keys and columnsOUTPUT
497097
"Xkr4" Check feature types
R
table(rowranges$gbkey)
OUTPUT
C_region D_segment exon J_segment misc_RNA
20 23 4008 94 1988
mRNA ncRNA precursor_RNA rRNA tRNA
21198 12285 1187 35 413
V_segment
535 Assemble SummarizedExperiment
R
stopifnot(rownames(rowranges) == rownames(counts),
rownames(coldata) == colnames(counts))
se <- SummarizedExperiment(
assays = list(counts = as.matrix(counts)),
rowRanges = as(rowranges, "GRanges"),
colData = coldata
)
Save SummarizedExperiment
R
saveRDS(se, "data/GSE96870_se.rds")
Session info
R
sessionInfo()
OUTPUT
R version 4.2.2 Patched (2022-11-10 r83330)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 22.04.2 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.10.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.10.0
locale:
[1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
[4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
[7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] org.Mm.eg.db_3.16.0 AnnotationDbi_1.60.0
[3] SummarizedExperiment_1.28.0 Biobase_2.58.0
[5] MatrixGenerics_1.10.0 matrixStats_0.63.0
[7] GenomicRanges_1.50.2 GenomeInfoDb_1.34.9
[9] IRanges_2.32.0 S4Vectors_0.36.1
[11] BiocGenerics_0.44.0
loaded via a namespace (and not attached):
[1] Rcpp_1.0.10 compiler_4.2.2 BiocManager_1.30.19
[4] XVector_0.38.0 bitops_1.0-7 tools_4.2.2
[7] zlibbioc_1.44.0 bit_4.0.5 RSQLite_2.2.20
[10] evaluate_0.20 memoise_2.0.1 lattice_0.20-45
[13] pkgconfig_2.0.3 png_0.1-8 rlang_1.0.6
[16] Matrix_1.5-3 DelayedArray_0.24.0 DBI_1.1.3
[19] cli_3.6.0 xfun_0.37 fastmap_1.1.0
[22] GenomeInfoDbData_1.2.9 httr_1.4.4 knitr_1.42
[25] Biostrings_2.66.0 vctrs_0.5.2 bit64_4.0.5
[28] grid_4.2.2 R6_2.5.1 blob_1.2.3
[31] KEGGREST_1.38.0 renv_0.17.0-38 RCurl_1.98-1.10
[34] cachem_1.0.6 crayon_1.5.2 Content from Exploratory analysis and quality control
Last updated on 2023-03-14 | Edit this page
Overview
Questions
Objectives
- Learn how to explore the gene expression matrix and perform common quality control steps.
- Learn how to set up an interactive application for exploratory analysis.
R
suppressPackageStartupMessages({
library(SummarizedExperiment)
library(DESeq2)
library(vsn)
library(ggplot2)
library(ComplexHeatmap)
library(RColorBrewer)
library(hexbin)
})
R
se <- readRDS("data/GSE96870_se.rds")
Exploratory analysis is crucial for quality control and to get to know our data. It can help us detect quality problems, sample swaps and contamination, as well as give us a sense of the most salient patterns present in the data. In this episode, we will learn about two common ways of performing exploratory analysis for RNA-seq data; namely clustering and principal component analysis (PCA). These tools are in no way limited to (or developed for) analysis of RNA-seq data. However, there are certain characteristics of count assays that need to be taken into account when they are applied to this type of data.
R
se <- se[rowSums(assay(se, "counts")) > 5, ]
dds <- DESeq2::DESeqDataSet(se[, se$tissue == "Cerebellum"],
design = ~ sex + time)
WARNING
Warning in DESeq2::DESeqDataSet(se[, se$tissue == "Cerebellum"], design = ~sex
+ : some variables in design formula are characters, converting to factorsLibrary size differences
R
ggplot(data.frame(sample = colnames(dds),
libSize = colSums(assay(dds, "counts"))),
aes(x = sample, y = libSize)) +
geom_bar(stat = "identity") + theme_bw() +
labs(x = "Sample", y = "Total count") +
theme(axis.text.x = element_text(angle = 90, hjust = 1, vjust = 0.5))
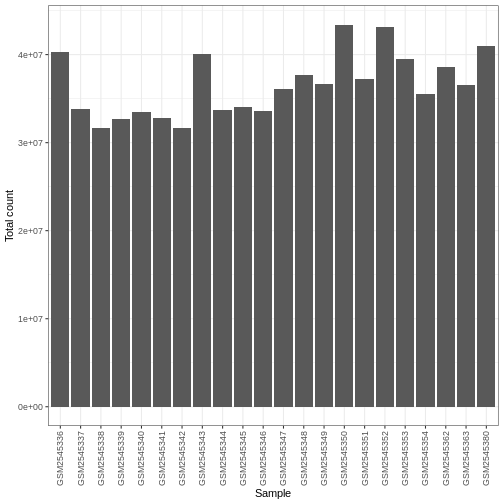
Differences in the total number of reads assigned to genes between samples typically occur for technical reasons. In practice, it means that we can not simply compare the raw read count directly between samples and conclude that a sample with a higher read count also expresses the gene more strongly - the higher count may be caused by an overall higher number of reads in that sample. In the rest of this section, we will use the term library size to refer to the total number of reads assigned to genes for a sample. We need to adjust for the differences in library size between samples, to avoid drawing incorrect conclusions. The way this is typically done for RNA-seq data can be described as a two-step procedure. First, we estimate size factors - sample-specific correction factors such that if the raw counts were to be divided by these factors, the resulting values would be more comparable across samples. Next, these size factors are incorporated into the statistical analysis of the data. It is important to pay close attention to how this is done in practice for a given analysis method. Sometimes the division of the counts by the size factors needs to be done explicitly by the analyst. Other times (as we will see for the differential expression analysis) it is important that they are provided separately to the analysis tool, which will then use them appropriately in the statistical model.
With DESeq2, size factors are calculated using the
estimateSizeFactors() function. The size factors estimated
by this function combines an adjustment for differences in library sizes
with an adjustment for differences in the RNA composition of the
samples. The latter is important due to the compositional nature of
RNA-seq data. There is a fixed number of reads to distribute between the
genes, and if a single (or a few) very highly expressed gene consume a
large part of the reads, all other genes will consequently receive very
low counts.
R
dds <- estimateSizeFactors(dds)
ggplot(data.frame(libSize = colSums(assay(dds, "counts")),
sizeFactor = sizeFactors(dds)),
aes(x = libSize, y = sizeFactor)) +
geom_point(size = 5) + theme_bw() +
labs(x = "Library size", y = "Size factor")
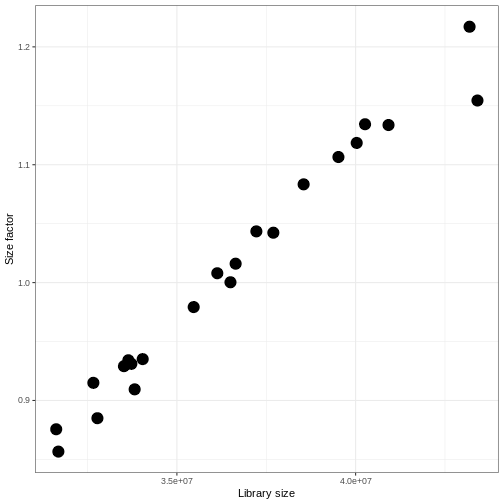
Transform data
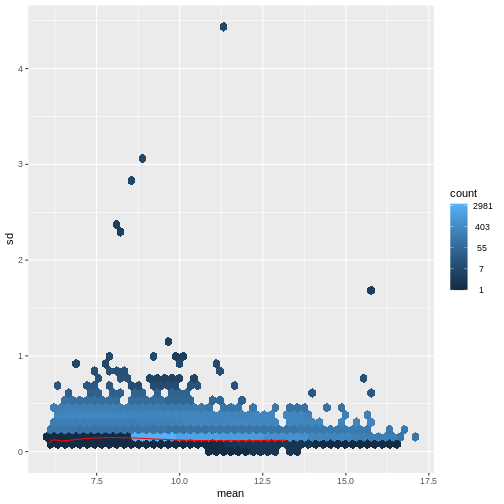
There is a rich literature on methods for exploratory analysis. Most of these work best in situations where the variance of the input data (here, each gene) is relatively independent of the average value. For read count data such as RNA-seq, this is not the case. In fact, the variance increases with the average read count.
R
meanSdPlot(assay(dds), ranks = FALSE)
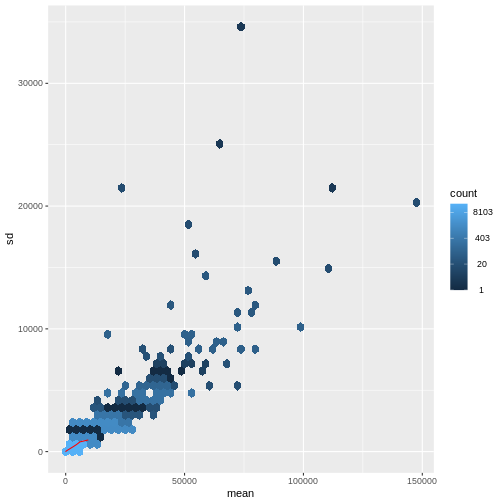
There are two ways around this: either we develop methods specifically adapted to count data, or we adapt (transform) the count data so that the existing methods are applicable. Both ways have been explored; however, at the moment the second approach is arguably more widely applied in practice.
R
vsd <- DESeq2::vst(dds, blind = TRUE)
meanSdPlot(assay(vsd), ranks = FALSE)
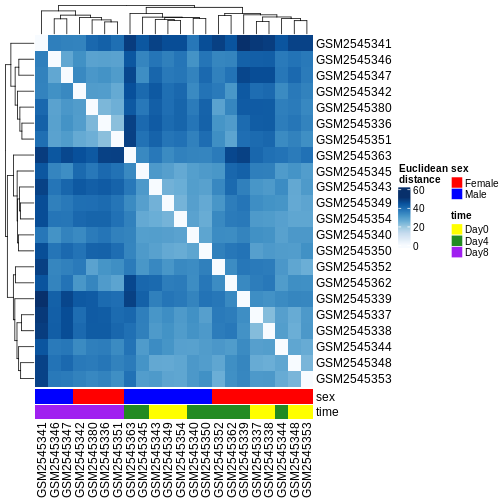
Heatmaps and clustering
R
dst <- dist(t(assay(vsd)))
colors <- colorRampPalette(brewer.pal(9, "Blues"))(255)
ComplexHeatmap::Heatmap(
as.matrix(dst),
col = colors,
name = "Euclidean\ndistance",
cluster_rows = hclust(dst),
cluster_columns = hclust(dst),
bottom_annotation = columnAnnotation(
sex = vsd$sex,
time = vsd$time,
col = list(sex = c(Female = "red", Male = "blue"),
time = c(Day0 = "yellow", Day4 = "forestgreen", Day8 = "purple")))
)
PCA
Principal component analysis is a dimensionality reduction method, which projects the samples into a lower-dimensional space. This lower-dimensional representation can be used for visualization, or as the input for other analysis methods. The principal components are defined in such a way that they are orthogonal, and that the projection of the samples into the space they span contains as much variance as possible. It is an unsupervised method in the sense that no external information about the samples (e.g., the treatment condition) is taken into account. In the plot below we represent the samples in a two-dimensional principal component space. For each of the two dimensions, we indicate the fraction of the total variance that is represented by that component. By definition, the first principal component will always represent more of the variance than the subsequent ones. The fraction of explained variance is a measure of how much of the ‘signal’ in the data that is retained when we project the samples from the original, high-dimensional space to the low-dimensional space for visualization.
R
pcaData <- DESeq2::plotPCA(vsd, intgroup = c("sex", "time"),
returnData = TRUE)
percentVar <- round(100 * attr(pcaData, "percentVar"))
ggplot(pcaData, aes(x = PC1, y = PC2)) +
geom_point(aes(color = sex, shape = time), size = 5) +
theme_minimal() +
xlab(paste0("PC1: ", percentVar[1], "% variance")) +
ylab(paste0("PC2: ", percentVar[2], "% variance")) +
coord_fixed() +
scale_color_manual(values = c(Male = "blue", Female = "red"))
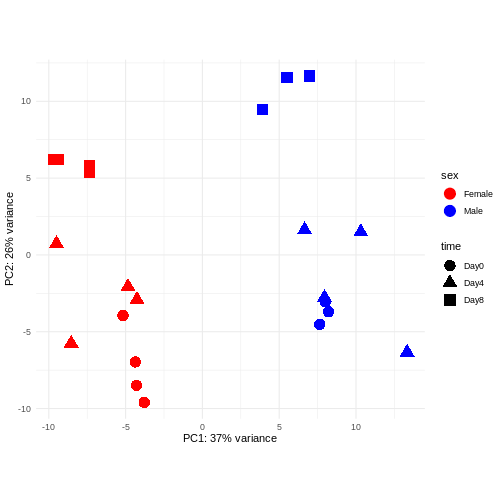
Session info
R
sessionInfo()
OUTPUT
R version 4.2.2 Patched (2022-11-10 r83330)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 22.04.2 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.10.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.10.0
locale:
[1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
[4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
[7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] hexbin_1.28.2 RColorBrewer_1.1-3
[3] ComplexHeatmap_2.14.0 ggplot2_3.4.0
[5] vsn_3.66.0 DESeq2_1.38.3
[7] SummarizedExperiment_1.28.0 Biobase_2.58.0
[9] MatrixGenerics_1.10.0 matrixStats_0.63.0
[11] GenomicRanges_1.50.2 GenomeInfoDb_1.34.9
[13] IRanges_2.32.0 S4Vectors_0.36.1
[15] BiocGenerics_0.44.0
loaded via a namespace (and not attached):
[1] httr_1.4.4 bit64_4.0.5 foreach_1.5.2
[4] highr_0.10 BiocManager_1.30.19 affy_1.76.0
[7] blob_1.2.3 renv_0.17.0-38 GenomeInfoDbData_1.2.9
[10] pillar_1.8.1 RSQLite_2.2.20 lattice_0.20-45
[13] glue_1.6.2 limma_3.54.1 digest_0.6.31
[16] XVector_0.38.0 colorspace_2.1-0 preprocessCore_1.60.2
[19] Matrix_1.5-3 XML_3.99-0.13 pkgconfig_2.0.3
[22] GetoptLong_1.0.5 zlibbioc_1.44.0 xtable_1.8-4
[25] scales_1.2.1 affyio_1.68.0 BiocParallel_1.32.5
[28] tibble_3.1.8 annotate_1.76.0 KEGGREST_1.38.0
[31] farver_2.1.1 generics_0.1.3 cachem_1.0.6
[34] withr_2.5.0 cli_3.6.0 magrittr_2.0.3
[37] crayon_1.5.2 memoise_2.0.1 evaluate_0.20
[40] fansi_1.0.4 doParallel_1.0.17 tools_4.2.2
[43] GlobalOptions_0.1.2 lifecycle_1.0.3 munsell_0.5.0
[46] locfit_1.5-9.7 cluster_2.1.4 DelayedArray_0.24.0
[49] AnnotationDbi_1.60.0 Biostrings_2.66.0 compiler_4.2.2
[52] rlang_1.0.6 RCurl_1.98-1.10 iterators_1.0.14
[55] circlize_0.4.15 rjson_0.2.21 labeling_0.4.2
[58] bitops_1.0-7 gtable_0.3.1 codetools_0.2-19
[61] DBI_1.1.3 R6_2.5.1 knitr_1.42
[64] dplyr_1.1.0 fastmap_1.1.0 bit_4.0.5
[67] utf8_1.2.3 clue_0.3-64 shape_1.4.6
[70] parallel_4.2.2 Rcpp_1.0.10 vctrs_0.5.2
[73] geneplotter_1.76.0 png_0.1-8 tidyselect_1.2.0
[76] xfun_0.37 Content from Differential expression analysis
Last updated on 2023-03-14 | Edit this page
Overview
Questions
- How do we find differentially expressed genes?
Objectives
- Explain the steps involved in a differential expression analysis.
- Explain how to perform these steps in R, using DESeq2.
R
suppressPackageStartupMessages({
library(SummarizedExperiment)
library(DESeq2)
library(ggplot2)
library(ExploreModelMatrix)
library(cowplot)
library(ComplexHeatmap)
})
Load data
R
se <- readRDS("data/GSE96870_se.rds")
Create DESeqDataSet
R
dds <- DESeq2::DESeqDataSet(se[, se$tissue == "Cerebellum"],
design = ~ sex + time)
WARNING
Warning in DESeq2::DESeqDataSet(se[, se$tissue == "Cerebellum"], design = ~sex
+ : some variables in design formula are characters, converting to factorsRun DESeq()
R
dds <- DESeq2::DESeq(dds)
OUTPUT
estimating size factorsOUTPUT
estimating dispersionsOUTPUT
gene-wise dispersion estimatesOUTPUT
mean-dispersion relationshipOUTPUT
final dispersion estimatesOUTPUT
fitting model and testingR
plotDispEsts(dds)
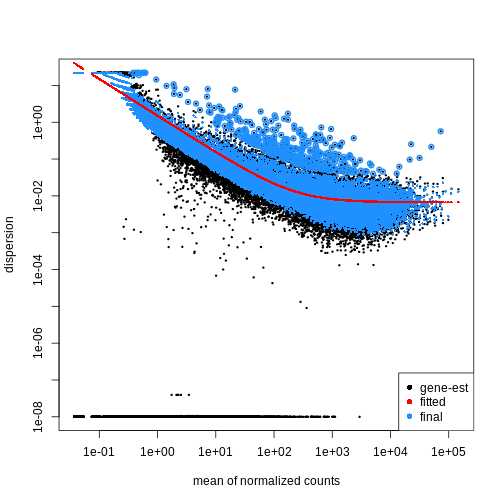
Extract results for specific contrasts
R
## Day 8 vs Day 0
resTime <- DESeq2::results(dds, contrast = c("time", "Day8", "Day0"))
summary(resTime)
OUTPUT
out of 32652 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 4472, 14%
LFC < 0 (down) : 4276, 13%
outliers [1] : 10, 0.031%
low counts [2] : 8732, 27%
(mean count < 1)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?resultsR
head(resTime[order(resTime$pvalue), ])
OUTPUT
log2 fold change (MLE): time Day8 vs Day0
Wald test p-value: time Day8 vs Day0
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
Asl 701.343 1.11733 0.0592541 18.8565 2.59885e-79
Apod 18765.146 1.44698 0.0805186 17.9708 3.30147e-72
Cyp2d22 2550.480 0.91020 0.0554756 16.4072 1.69794e-60
Klk6 546.503 -1.67190 0.1058989 -15.7877 3.78228e-56
Fcrls 184.235 -1.94701 0.1279847 -15.2128 2.90708e-52
A330076C08Rik 107.250 -1.74995 0.1154279 -15.1606 6.45112e-52
padj
<numeric>
Asl 6.21386e-75
Apod 3.94690e-68
Cyp2d22 1.35326e-56
Klk6 2.26086e-52
Fcrls 1.39017e-48
A330076C08Rik 2.57077e-48R
DESeq2::plotMA(resTime)
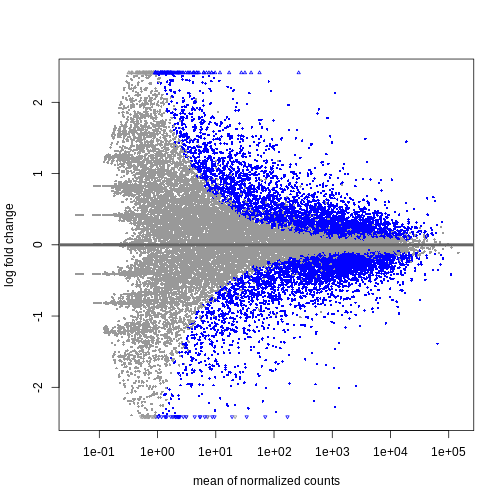
R
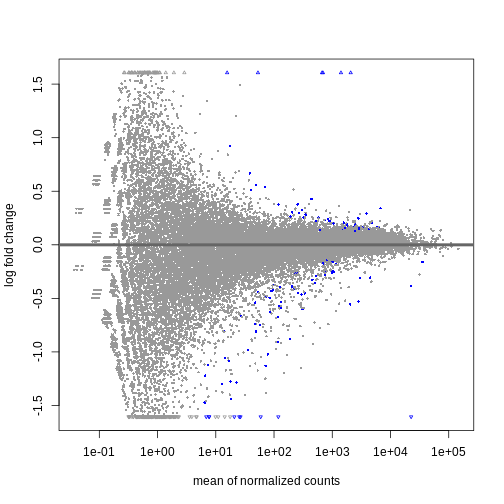
## Male vs Female
resSex <- DESeq2::results(dds, contrast = c("sex", "Male", "Female"))
summary(resSex)
OUTPUT
out of 32652 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 53, 0.16%
LFC < 0 (down) : 71, 0.22%
outliers [1] : 10, 0.031%
low counts [2] : 13717, 42%
(mean count < 6)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?resultsR
head(resSex[order(resSex$pvalue), ])
OUTPUT
log2 fold change (MLE): sex Male vs Female
Wald test p-value: sex Male vs Female
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
Xist 22603.0359 -11.60429 0.336282 -34.5076 6.16852e-261
Ddx3y 2072.9436 11.87241 0.397493 29.8683 5.08722e-196
Eif2s3y 1410.8750 12.62514 0.565216 22.3369 1.62066e-110
Kdm5d 692.1672 12.55386 0.593627 21.1477 2.89566e-99
Uty 667.4375 12.01728 0.593591 20.2451 3.92780e-91
LOC105243748 52.9669 9.08325 0.597624 15.1989 3.59432e-52
padj
<numeric>
Xist 1.16739e-256
Ddx3y 4.81378e-192
Eif2s3y 1.02237e-106
Kdm5d 1.37001e-95
Uty 1.48667e-87
LOC105243748 1.13371e-48R
DESeq2::plotMA(resSex)
Visualize selected set of genes
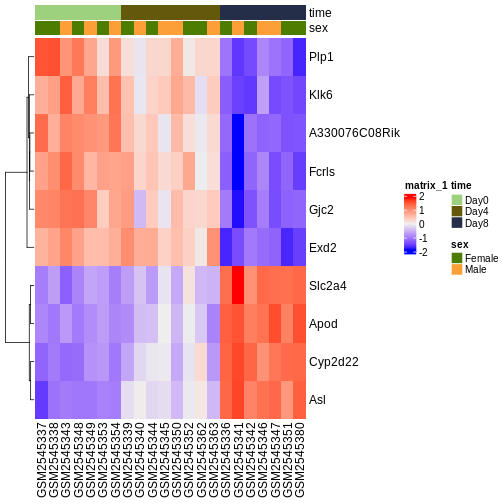
R
vsd <- DESeq2::vst(dds, blind = TRUE)
genes <- rownames(head(resTime[order(resTime$pvalue), ], 10))
heatmapData <- assay(vsd)[genes, ]
heatmapData <- t(scale(t(heatmapData)))
heatmapColAnnot <- data.frame(colData(vsd)[, c("time", "sex")])
idx <- order(vsd$time)
heatmapData <- heatmapData[, idx]
heatmapColAnnot <- HeatmapAnnotation(df = heatmapColAnnot[idx, ])
ComplexHeatmap::Heatmap(heatmapData,
top_annotation = heatmapColAnnot,
cluster_rows = TRUE, cluster_columns = FALSE)
Content from Gene set analysis
Last updated on 2023-03-14 | Edit this page
Overview
Questions
- How do we find differentially expressed pathways?
Objectives
- Explain how to find differentially expressed pathways with gene set analysis in R.
- Understand how differentially expressed genes can enrich a gene set.
- Explain how to perform a gene set analysis in R, using clusterProfiler.
First, we are going to explore the basic concept of enriching a gene set with differentially expressed (DE) genes. Recall the differential expression analysis.
R
library(SummarizedExperiment)
library(DESeq2)
R
se <- readRDS("data/GSE96870_se.rds")
R
dds <- DESeq2::DESeqDataSet(se[, se$tissue == "Cerebellum"],
design = ~ sex + time)
WARNING
Warning in DESeq2::DESeqDataSet(se[, se$tissue == "Cerebellum"], design = ~sex
+ : some variables in design formula are characters, converting to factorsR
dds <- DESeq2::DESeq(dds)
Fetch results for the contrast between male and female mice.
R
resSex <- DESeq2::results(dds, contrast = c("sex", "Male", "Female"))
summary(resSex)
OUTPUT
out of 32652 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 53, 0.16%
LFC < 0 (down) : 71, 0.22%
outliers [1] : 10, 0.031%
low counts [2] : 13717, 42%
(mean count < 6)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?resultsSelect DE genes between males and females with FDR < 5%.
R
sexDE <- as.data.frame(subset(resSex, padj < 0.05))
dim(sexDE)
OUTPUT
[1] 54 6R
sexDE <- sexDE[order(abs(sexDE$log2FoldChange), decreasing=TRUE), ]
head(sexDE)
OUTPUT
baseMean log2FoldChange lfcSE stat pvalue
Eif2s3y 1410.8750 12.62514 0.5652155 22.33685 1.620659e-110
Kdm5d 692.1672 12.55386 0.5936267 21.14773 2.895664e-99
Uty 667.4375 12.01728 0.5935911 20.24505 3.927797e-91
Ddx3y 2072.9436 11.87241 0.3974927 29.86825 5.087220e-196
Xist 22603.0359 -11.60429 0.3362822 -34.50761 6.168523e-261
LOC105243748 52.9669 9.08325 0.5976242 15.19893 3.594320e-52
padj
Eif2s3y 1.022366e-106
Kdm5d 1.370011e-95
Uty 1.486671e-87
Ddx3y 4.813782e-192
Xist 1.167393e-256
LOC105243748 1.133708e-48R
sexDEgenes <- rownames(sexDE)
head(sexDEgenes)
OUTPUT
[1] "Eif2s3y" "Kdm5d" "Uty" "Ddx3y" "Xist"
[6] "LOC105243748"R
length(sexDEgenes)
OUTPUT
[1] 54Enrichment of a curated gene set
Contribute!
Here we illustrate how to assess the enrichment of one gene set we curate ourselves with our subset of DE genes with sex-specific expression. Here we form such a gene set with genes from sex chromosomes. Could you think of another more accurate gene set formed by genes with sex-specific expression?
Build a gene set formed by genes located in the sex chromosomes X and Y.
R
xygenes <- rownames(se)[decode(seqnames(rowRanges(se)) %in% c("X", "Y"))]
length(xygenes)
OUTPUT
[1] 2123Build a contingency table and conduct a one-tailed Fisher’s exact test that verifies the association between genes being DE between male and female mice and being located in a sex chromosome.
R
N <- nrow(se)
n <- length(sexDEgenes)
m <- length(xygenes)
k <- length(intersect(xygenes, sexDEgenes))
dnames <- list(GS=c("inside", "outside"), DE=c("yes", "no"))
t <- matrix(c(k, n-k, m-k, N+k-n-m),
nrow=2, ncol=2, dimnames=dnames)
t
OUTPUT
DE
GS yes no
inside 18 2105
outside 36 39627R
fisher.test(t, alternative="greater")
OUTPUT
Fisher's Exact Test for Count Data
data: t
p-value = 7.944e-11
alternative hypothesis: true odds ratio is greater than 1
95 percent confidence interval:
5.541517 Inf
sample estimates:
odds ratio
9.411737 Gene ontology analysis with clusterProfiler
Contribute!
Here we illustrate how to assess the enrichment on the entire collection of Gene Ontology (GO) gene sets using the package clusterProfiler. Could we illustrate any missing important feature of this package for this analysis objective? Could we briefly mention other packages that may be useful for this task?
Second, let’s perform a gene set analysis for an entire collection of gene sets using the Bioconductor package clusterProfiler. For this purpose, we will fetch the results for the contrast between two time points.
R
resTime <- DESeq2::results(dds, contrast = c("time", "Day8", "Day0"))
summary(resTime)
OUTPUT
out of 32652 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 4472, 14%
LFC < 0 (down) : 4276, 13%
outliers [1] : 10, 0.031%
low counts [2] : 8732, 27%
(mean count < 1)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?resultsSelect DE genes between Day0 and Day8 with
FDR < 5% and minimum 1.5-fold change.
R
timeDE <- as.data.frame(subset(resTime, padj < 0.05 & abs(log2FoldChange) > log2(1.5)))
dim(timeDE)
OUTPUT
[1] 2110 6R
timeDE <- timeDE[order(abs(timeDE$log2FoldChange), decreasing=TRUE), ]
head(timeDE)
OUTPUT
baseMean log2FoldChange lfcSE stat pvalue
LOC105245444 2.441873 4.768938 0.9013067 5.291138 1.215573e-07
LOC105246405 9.728219 4.601505 0.6101832 7.541186 4.657174e-14
4933427D06Rik 1.480365 4.556126 1.0318402 4.415535 1.007607e-05
A930006I01Rik 2.312732 -4.353155 0.9176026 -4.744053 2.094837e-06
LOC105245223 3.272536 4.337202 0.8611255 5.036666 4.737099e-07
A530053G22Rik 1.554735 4.243903 1.0248977 4.140806 3.460875e-05
padj
LOC105245444 1.800765e-06
LOC105246405 2.507951e-12
4933427D06Rik 9.169093e-05
A930006I01Rik 2.252139e-05
LOC105245223 6.047199e-06
A530053G22Rik 2.720142e-04R
timeDEgenes <- rownames(timeDE)
head(timeDEgenes)
OUTPUT
[1] "LOC105245444" "LOC105246405" "4933427D06Rik" "A930006I01Rik"
[5] "LOC105245223" "A530053G22Rik"R
length(timeDEgenes)
OUTPUT
[1] 2110Call the enrichGO() function from clusterProfiler
as follows.
R
library(clusterProfiler)
library(org.Mm.eg.db)
library(enrichplot)
resTimeGO <- enrichGO(gene = timeDEgenes,
keyType = "SYMBOL",
universe = rownames(se),
OrgDb = org.Mm.eg.db,
ont = "BP",
pvalueCutoff = 0.01,
qvalueCutoff = 0.01)
dim(resTimeGO)
OUTPUT
[1] 17 9R
head(resTimeGO)
OUTPUT
ID Description
GO:0035456 GO:0035456 response to interferon-beta
GO:0071674 GO:0071674 mononuclear cell migration
GO:0035458 GO:0035458 cellular response to interferon-beta
GO:0050900 GO:0050900 leukocyte migration
GO:0030595 GO:0030595 leukocyte chemotaxis
GO:0002523 GO:0002523 leukocyte migration involved in inflammatory response
GeneRatio BgRatio pvalue p.adjust qvalue
GO:0035456 17/1260 54/21417 6.434058e-09 3.254347e-05 3.082930e-05
GO:0071674 32/1260 179/21417 1.596474e-08 4.037482e-05 3.824814e-05
GO:0035458 15/1260 47/21417 4.064983e-08 6.749342e-05 6.393832e-05
GO:0050900 50/1260 375/21417 5.337558e-08 6.749342e-05 6.393832e-05
GO:0030595 34/1260 222/21417 2.880367e-07 2.913780e-04 2.760302e-04
GO:0002523 10/1260 25/21417 6.944905e-07 5.854555e-04 5.546176e-04
geneID
GO:0035456 Tgtp1/Tgtp2/F830016B08Rik/Iigp1/Ifitm6/Igtp/Gm4951/Bst2/Oas1c/Irgm1/Gbp6/Ifi47/Aim2/Ifitm7/Irgm2/Ifit1/Ifi204
GO:0071674 Tnfsf18/Aire/Ccl17/Ccr7/Nlrp12/Ccl2/Retnlg/Apod/Il12a/Ccl5/Fut7/Ccl7/Spn/Itgb3/Grem1/Ptk2b/Lgals3/Adam8/Dusp1/Ch25h/Nbl1/Alox5/Padi2/Plg/Calr/Ager/Slamf9/Ccl6/Mdk/Itga4/Hsd3b7/Trpm4
GO:0035458 Tgtp1/Tgtp2/F830016B08Rik/Iigp1/Igtp/Gm4951/Oas1c/Irgm1/Gbp6/Ifi47/Aim2/Ifitm7/Irgm2/Ifit1/Ifi204
GO:0050900 Tnfsf18/Aire/Ccl17/Ccr7/Nlrp12/Bst1/Ccl2/Retnlg/Ppbp/Cxcl5/Apod/Il12a/Ccl5/Umodl1/Fut7/Ccl7/Ccl28/Spn/Sell/Itgb3/Grem1/Cxcl1/Ptk2b/Lgals3/Adam8/Pf4/Dusp1/Ch25h/S100a8/Nbl1/Alox5/Padi2/Plg/Edn3/Il33/Ptn/Ada/Enpp1/Calr/Ager/Slamf9/Ccl6/Prex1/Aoc3/Itgam/Mdk/Itga4/Hsd3b7/P2ry12/Trpm4
GO:0030595 Tnfsf18/Ccl17/Ccr7/Bst1/Ccl2/Retnlg/Ppbp/Cxcl5/Il12a/Ccl5/Ccl7/Sell/Grem1/Cxcl1/Ptk2b/Lgals3/Adam8/Pf4/Dusp1/Ch25h/S100a8/Nbl1/Alox5/Padi2/Edn3/Ptn/Calr/Slamf9/Ccl6/Prex1/Itgam/Mdk/Hsd3b7/Trpm4
GO:0002523 Ccl2/Ppbp/Fut7/Adam8/S100a8/Alox5/Ptn/Aoc3/Itgam/Mdk
Count
GO:0035456 17
GO:0071674 32
GO:0035458 15
GO:0050900 50
GO:0030595 34
GO:0002523 10Let’s build a more readable table of results.
R
library(kableExtra)
resTimeGOtab <- as.data.frame(resTimeGO)
resTimeGOtab$ID <- NULL
resTimeGOtab$geneID <- sapply(strsplit(resTimeGO$geneID, "/"), paste, collapse=", ")
ktab <- kable(resTimeGOtab, row.names=TRUE, caption="GO results for DE genes between time points.")
kable_styling(ktab, bootstrap_options=c("stripped", "hover", "responsive"), fixed_thead=TRUE)
| Description | GeneRatio | BgRatio | pvalue | p.adjust | qvalue | geneID | Count | |
|---|---|---|---|---|---|---|---|---|
| GO:0035456 | response to interferon-beta | 17/1260 | 54/21417 | 0.00e+00 | 0.0000325 | 0.0000308 | Tgtp1, Tgtp2, F830016B08Rik, Iigp1, Ifitm6, Igtp, Gm4951, Bst2, Oas1c, Irgm1, Gbp6, Ifi47, Aim2, Ifitm7, Irgm2, Ifit1, Ifi204 | 17 |
| GO:0071674 | mononuclear cell migration | 32/1260 | 179/21417 | 0.00e+00 | 0.0000404 | 0.0000382 | Tnfsf18, Aire, Ccl17, Ccr7, Nlrp12, Ccl2, Retnlg, Apod, Il12a, Ccl5, Fut7, Ccl7, Spn, Itgb3, Grem1, Ptk2b, Lgals3, Adam8, Dusp1, Ch25h, Nbl1, Alox5, Padi2, Plg, Calr, Ager, Slamf9, Ccl6, Mdk, Itga4, Hsd3b7, Trpm4 | 32 |
| GO:0035458 | cellular response to interferon-beta | 15/1260 | 47/21417 | 0.00e+00 | 0.0000675 | 0.0000639 | Tgtp1, Tgtp2, F830016B08Rik, Iigp1, Igtp, Gm4951, Oas1c, Irgm1, Gbp6, Ifi47, Aim2, Ifitm7, Irgm2, Ifit1, Ifi204 | 15 |
| GO:0050900 | leukocyte migration | 50/1260 | 375/21417 | 1.00e-07 | 0.0000675 | 0.0000639 | Tnfsf18, Aire, Ccl17, Ccr7, Nlrp12, Bst1, Ccl2, Retnlg, Ppbp, Cxcl5, Apod, Il12a, Ccl5, Umodl1, Fut7, Ccl7, Ccl28, Spn, Sell, Itgb3, Grem1, Cxcl1, Ptk2b, Lgals3, Adam8, Pf4, Dusp1, Ch25h, S100a8, Nbl1, Alox5, Padi2, Plg, Edn3, Il33, Ptn, Ada, Enpp1, Calr, Ager, Slamf9, Ccl6, Prex1, Aoc3, Itgam, Mdk, Itga4, Hsd3b7, P2ry12, Trpm4 | 50 |
| GO:0030595 | leukocyte chemotaxis | 34/1260 | 222/21417 | 3.00e-07 | 0.0002914 | 0.0002760 | Tnfsf18, Ccl17, Ccr7, Bst1, Ccl2, Retnlg, Ppbp, Cxcl5, Il12a, Ccl5, Ccl7, Sell, Grem1, Cxcl1, Ptk2b, Lgals3, Adam8, Pf4, Dusp1, Ch25h, S100a8, Nbl1, Alox5, Padi2, Edn3, Ptn, Calr, Slamf9, Ccl6, Prex1, Itgam, Mdk, Hsd3b7, Trpm4 | 34 |
| GO:0002523 | leukocyte migration involved in inflammatory response | 10/1260 | 25/21417 | 7.00e-07 | 0.0005855 | 0.0005546 | Ccl2, Ppbp, Fut7, Adam8, S100a8, Alox5, Ptn, Aoc3, Itgam, Mdk | 10 |
| GO:0050953 | sensory perception of light stimulus | 26/1260 | 162/21417 | 2.90e-06 | 0.0020909 | 0.0019808 | Aipl1, Vsx2, Nxnl2, Lrit3, Cryba2, Bfsp2, Lrat, Gabrr2, Lum, Rlbp1, Pde6g, Gpr179, Col1a1, Cplx3, Best1, Ush1g, Rs1, Rdh5, Guca1b, Th, Ppef2, Rbp4, Olfm2, Rom1, Vsx1, Rpe65 | 26 |
| GO:0071675 | regulation of mononuclear cell migration | 21/1260 | 117/21417 | 4.20e-06 | 0.0026851 | 0.0025436 | Tnfsf18, Aire, Ccr7, Ccl2, Apod, Il12a, Ccl5, Ccl7, Spn, Itgb3, Grem1, Ptk2b, Lgals3, Adam8, Dusp1, Nbl1, Padi2, Calr, Ager, Mdk, Itga4 | 21 |
| GO:0007601 | visual perception | 25/1260 | 158/21417 | 5.80e-06 | 0.0032508 | 0.0030795 | Aipl1, Vsx2, Nxnl2, Lrit3, Cryba2, Bfsp2, Lrat, Gabrr2, Lum, Rlbp1, Pde6g, Gpr179, Col1a1, Cplx3, Best1, Rs1, Rdh5, Guca1b, Th, Ppef2, Rbp4, Olfm2, Rom1, Vsx1, Rpe65 | 25 |
| GO:0034341 | response to interferon-gamma | 22/1260 | 135/21417 | 1.28e-05 | 0.0056936 | 0.0053937 | Ccl17, Gbp4, Ccl2, Tgtp1, H2-Q7, Il12rb1, Ifitm6, Ccl5, Ccl7, Igtp, Nos2, Nlrc5, Bst2, Irgm1, Gbp6, Capg, Ifitm7, Gbp9, Gbp5, Irgm2, Ccl6, Aqp4 | 22 |
| GO:0060326 | cell chemotaxis | 38/1260 | 308/21417 | 1.30e-05 | 0.0056936 | 0.0053937 | Tnfsf18, Ccl17, Ccr7, Bst1, Ccl2, Retnlg, Ppbp, Cxcl5, Nr4a1, Il12a, Ccl5, Ccl7, Ccl28, Sell, Grem1, Cxcl1, Ptk2b, Lgals3, Adam8, Pf4, Dusp1, Ch25h, S100a8, Nbl1, Alox5, Padi2, Edn3, Ptn, Plxnb3, Calr, Lpar1, Slamf9, Ccl6, Prex1, Itgam, Mdk, Hsd3b7, Trpm4 | 38 |
| GO:0002685 | regulation of leukocyte migration | 31/1260 | 230/21417 | 1.41e-05 | 0.0056936 | 0.0053937 | Tnfsf18, Aire, Ccr7, Bst1, Ccl2, Apod, Il12a, Ccl5, Fut7, Ccl7, Ccl28, Spn, Sell, Itgb3, Grem1, Ptk2b, Lgals3, Adam8, Dusp1, Nbl1, Padi2, Edn3, Il33, Ptn, Ada, Calr, Ager, Aoc3, Mdk, Itga4, P2ry12 | 31 |
| GO:0036336 | dendritic cell migration | 9/1260 | 27/21417 | 1.46e-05 | 0.0056936 | 0.0053937 | Tnfsf18, Ccr7, Nlrp12, Retnlg, Il12a, Alox5, Calr, Slamf9, Trpm4 | 9 |
| GO:1990266 | neutrophil migration | 21/1260 | 127/21417 | 1.59e-05 | 0.0057426 | 0.0054401 | Ccl17, Ccr7, Bst1, Ccl2, Ppbp, Cxcl5, Ccl5, Umodl1, Fut7, Ccl7, Sell, Cxcl1, Lgals3, Adam8, Pf4, S100a8, Edn3, Ccl6, Prex1, Itgam, Mdk | 21 |
| GO:0016264 | gap junction assembly | 7/1260 | 16/21417 | 1.71e-05 | 0.0057811 | 0.0054766 | Gjd3, Aplnr, Gja5, Agt, Hopx, Cav1, Ace | 7 |
| GO:0030593 | neutrophil chemotaxis | 18/1260 | 100/21417 | 1.94e-05 | 0.0061338 | 0.0058107 | Ccl17, Ccr7, Bst1, Ccl2, Ppbp, Cxcl5, Ccl5, Ccl7, Sell, Cxcl1, Lgals3, Pf4, S100a8, Edn3, Ccl6, Prex1, Itgam, Mdk | 18 |
| GO:1903028 | positive regulation of opsonization | 6/1260 | 12/21417 | 2.78e-05 | 0.0082825 | 0.0078462 | Pla2g5, Masp2, C4b, Colec11, C3, Cfp | 6 |