Differential expression analysis
Last updated on 2023-03-14 | Edit this page
Overview
Questions
- How do we find differentially expressed genes?
Objectives
- Explain the steps involved in a differential expression analysis.
- Explain how to perform these steps in R, using DESeq2.
R
suppressPackageStartupMessages({
library(SummarizedExperiment)
library(DESeq2)
library(ggplot2)
library(ExploreModelMatrix)
library(cowplot)
library(ComplexHeatmap)
})
Load data
R
se <- readRDS("data/GSE96870_se.rds")
Create DESeqDataSet
R
dds <- DESeq2::DESeqDataSet(se[, se$tissue == "Cerebellum"],
design = ~ sex + time)
WARNING
Warning in DESeq2::DESeqDataSet(se[, se$tissue == "Cerebellum"], design = ~sex
+ : some variables in design formula are characters, converting to factorsRun DESeq()
R
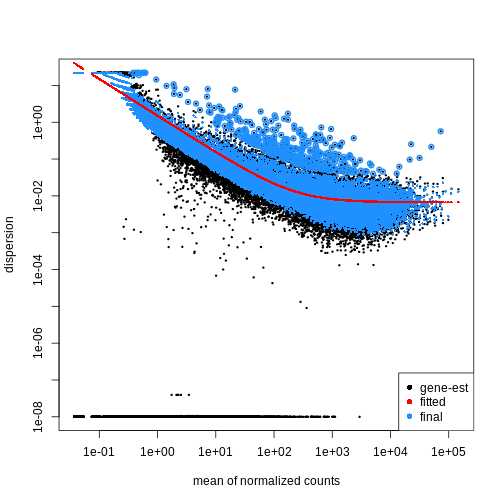
dds <- DESeq2::DESeq(dds)
OUTPUT
estimating size factorsOUTPUT
estimating dispersionsOUTPUT
gene-wise dispersion estimatesOUTPUT
mean-dispersion relationshipOUTPUT
final dispersion estimatesOUTPUT
fitting model and testingR
plotDispEsts(dds)
Extract results for specific contrasts
R
## Day 8 vs Day 0
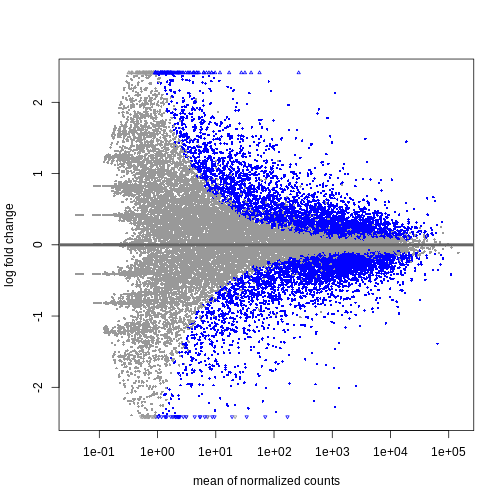
resTime <- DESeq2::results(dds, contrast = c("time", "Day8", "Day0"))
summary(resTime)
OUTPUT
out of 32652 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 4472, 14%
LFC < 0 (down) : 4276, 13%
outliers [1] : 10, 0.031%
low counts [2] : 8732, 27%
(mean count < 1)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?resultsR
head(resTime[order(resTime$pvalue), ])
OUTPUT
log2 fold change (MLE): time Day8 vs Day0
Wald test p-value: time Day8 vs Day0
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
Asl 701.343 1.11733 0.0592541 18.8565 2.59885e-79
Apod 18765.146 1.44698 0.0805186 17.9708 3.30147e-72
Cyp2d22 2550.480 0.91020 0.0554756 16.4072 1.69794e-60
Klk6 546.503 -1.67190 0.1058989 -15.7877 3.78228e-56
Fcrls 184.235 -1.94701 0.1279847 -15.2128 2.90708e-52
A330076C08Rik 107.250 -1.74995 0.1154279 -15.1606 6.45112e-52
padj
<numeric>
Asl 6.21386e-75
Apod 3.94690e-68
Cyp2d22 1.35326e-56
Klk6 2.26086e-52
Fcrls 1.39017e-48
A330076C08Rik 2.57077e-48R
DESeq2::plotMA(resTime)
R
## Male vs Female
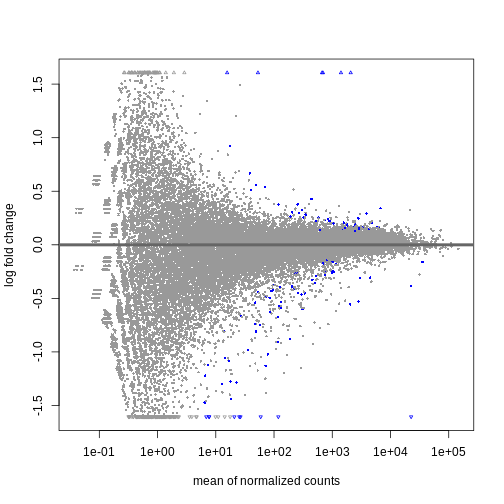
resSex <- DESeq2::results(dds, contrast = c("sex", "Male", "Female"))
summary(resSex)
OUTPUT
out of 32652 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 53, 0.16%
LFC < 0 (down) : 71, 0.22%
outliers [1] : 10, 0.031%
low counts [2] : 13717, 42%
(mean count < 6)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?resultsR
head(resSex[order(resSex$pvalue), ])
OUTPUT
log2 fold change (MLE): sex Male vs Female
Wald test p-value: sex Male vs Female
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
Xist 22603.0359 -11.60429 0.336282 -34.5076 6.16852e-261
Ddx3y 2072.9436 11.87241 0.397493 29.8683 5.08722e-196
Eif2s3y 1410.8750 12.62514 0.565216 22.3369 1.62066e-110
Kdm5d 692.1672 12.55386 0.593627 21.1477 2.89566e-99
Uty 667.4375 12.01728 0.593591 20.2451 3.92780e-91
LOC105243748 52.9669 9.08325 0.597624 15.1989 3.59432e-52
padj
<numeric>
Xist 1.16739e-256
Ddx3y 4.81378e-192
Eif2s3y 1.02237e-106
Kdm5d 1.37001e-95
Uty 1.48667e-87
LOC105243748 1.13371e-48R
DESeq2::plotMA(resSex)
Visualize selected set of genes
R
vsd <- DESeq2::vst(dds, blind = TRUE)
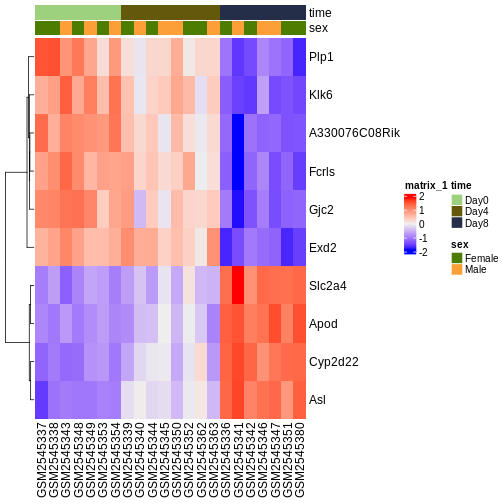
genes <- rownames(head(resTime[order(resTime$pvalue), ], 10))
heatmapData <- assay(vsd)[genes, ]
heatmapData <- t(scale(t(heatmapData)))
heatmapColAnnot <- data.frame(colData(vsd)[, c("time", "sex")])
idx <- order(vsd$time)
heatmapData <- heatmapData[, idx]
heatmapColAnnot <- HeatmapAnnotation(df = heatmapColAnnot[idx, ])
ComplexHeatmap::Heatmap(heatmapData,
top_annotation = heatmapColAnnot,
cluster_rows = TRUE, cluster_columns = FALSE)